Строение митохондрий биохимия
Строение, функции митохондрий
Митохондрии (греч. mhos— нить + chondros— зерно) — сферические или нитевидные органеллы шириной 0,5-1 мкм, длина которых может достигать 10 мкм. Они обычно накапливаются в тех участках цитоплазмы, где энергия используется наиболее интенсивно, как, например, в апикальных частях реснитчатых клеток, в среднем сегменте сперматозоидов или в базальной части клеток, переносящих ионы.
Эти органеллы преобразуют химическую энергию продуктов обмена, имеющихся в цитоплазме, в энергию, легкодоступную клетке. Около 50% этой энергии запасается в виде высокоэнергетических фосфатных связей в молекулах АТФ, а остальные 50% рассеиваются в видетепладля поддержания температуры тела.
Благодаря активности фермента АТФазы, АТФ быстро выделяет энергию, когда это необходимо клетке для выполнения любого типа работы — осмотической, механической, электрической или химической.
Митохондрии обладают характерной ультраструктурой. В их состав входят наружная и внутренняя митохондриальные мембраны; внутренняя мембрана образует складки, выступающие в глубь митохондрии, — кристы. Эти мембраны ограничивают два компартмента. Первый из них расположен между двумя мембранами — это межмембранное пространство.
Второй компартмент ограничен внутренней мембраной — это пространство между кристами, или пространство матрикса. По сравнению с другими мембранами клетки митохондриальные мембраны содержат очень большое количество белковых молекул. У большинства митохондрий кристы плоские и по форме сходны с выступающими внутрь полками, тогда как клетки, секретирующие стероиды (например, клетки надпочечников), часто содержат тубулярныс (имеющие вид трубочек) кристы.
Кристы увеличивают внутреннюю площадь поверхности митохондрии и содержат ферменты и другие компоненты систем окислительного фосфорилирования и транспорта электронов.
Система фосфорилирования аденозиндифосфата (АДФ) до АТФ локализована в глобулярных структурах, связанных с внутренней мембраной цилиндрическими ножками. Глобулярные структуры представляют собой комплекс белков с активностью АТФ-синтетазы, которая в присутствии АДФ, неорганического фосфата и энергии образует АТФ. Согласно хемиосмотической теории, синтез АТФ происходит за счет потока протонов через эту глобулярную структуру.

Число митохондрий и содержание крист в каждой митохондрии связаны с энергетической активностью той клетки, в которой они расположены. Так, клетки с высоким энергетическим обменом (например, клетки сердечной мышцы, клетки некоторых почечных канальцев) имеют многочисленные митохондрии с большим числом плотно упакованных крист, тогда как клетки с низким энергетическим обменом содержатлишь небольшое количество митохондрий с короткими кристами.
Между кристами располагается богатый белками аморфный матрикс, содержащий кольцевые молекулы ДНК и три варианта РНК. В клетках различных типов в митохондриальном матриксе выявляются также округлые электронно-плотные гранулы с высоким содержанием Са2+. Хотя функция этого катиона в митохондриях ясна не полностью, он может играть важную роль в регуляции активности некоторых митохондриальных ферментов; другая его функциональная роль связана с необходимостью поддержания низкой концентрации Са2+ в цитозоле.
Митохондрии закачивают внутрь себя Са2+ при его высоких концентрациях в цитозоле. В пространстве матрикса находятся ферменты цикла лимонной кислоты (цикла Крсбса) и бета-окисления жирных кислот. ДНК, выделенная из митохондриального матрикса, является двухцепочечной и имеет кольцевую структуру, очень сходную с таковой в хромосомах бактерий. Эти нити синтезируются в митохондрии; их редупликация протекает независимо от репликаци и ядерной ДНК. М итохондрии содержат три типа РНК: рибосомальную РНК (рРНК), информационную РНК(иРНК) и транспортную РНК (тРНК). Митохондриальные рибосомы имеют меньшие размеры, чем рибосомы, расположенные в цитозоле, и сходны с бактериальными рибосомами.
В митохондриях происходит белковый синтез, но из-за небольшого количества митохондриальной ДНК только малая доля митохондриальных белков вырабатывается местно. Большая же их часть кодируется ядерной ДНК и синтезируется на полирибосомах, расположенных в цитозоле. Эти белки содержат небольшую последовательность аминокислот, которая служит сигналом об их митохондриальном назначении, причем они транспортируются в митохондрии посредством механизма, потребляющего энергию.
Начальное расщепление углеводов и жиров происходит в цитоплазматическом матриксе. Конечным метаболическим продуктом этих внемитохондриальных путей обмена веществ является ацетилкоэнзим А, который далее попадает в митохондрии. Внутри митохондрий ацетилкоэнзим А взаимодействует с оксалоацетатом, образуя лимонную кислоту. В цикле лимонной кислоты в результате нескольких реакций декарбоксилирования образуется углекислый газ, а специфические реакции, катализируемые дегидрогеназой, приводят к удалению четырех пар ионов Н+.
Ионы Н+ в конечном итоге реагируют с кислородом, формируя воду. Благодаря активности цитохромов а, b и с, кофермента Q и цитохромоксидазы, электронтранспортная система, расположенная на внутренней митохондриальной мембране, выделяет энергию, которая захватывается в трех участках этой системы путем образования АТФ из АДФ и неорганического фосфата. В аэробных условиях сочетанная активность внемитохондриального гликолиза и цикла лимонной кислоты, а также электронтранспортной системы дает 36 молекул АТФ на одну молекулу глюкозы. Это в 18 раз превышает количество энергии, получаемое в анаэробных условиях, когда может использоваться только гликолитический путь.
В процессе митоза каждая дочерняя клетка получает примерно половину митохондрий, первоначально имевшихся в материнской клетке. Новые митохондрии образуются из ранее имевшихся посредством роста и последующего разделения (расщепления) самой органеллы.
Тот факт, что митохондрии обладают некоторыми характеристиками, общими с бактериями, привел к гипотезе, согласно которой митохондрии произошли из первоначально существовавших аэробных прокариот, адаптировавшихся к эндосимбиотической жизни внутри эукариотической клетки-хозяина (внутриклеточному симбиозу).
Описаны несколько болезней, обусловленных недостаточной деятельностью митохондрий, причем большинство из них характеризуется нарушением функции мышц. Вследствие высокой активности энергетического обмена к митохондриальным дефектам очень чувствительны волокна скелетных мышц. Мутации ДНК или дефекты, которые могут возникать в митохондриях или клеточном ядре, вызывают митохондриальные болезни.
Наследование митохондрий осуществляется по материнской линии, так как в цитоплазме зиготы митохондрии сперматозоида остаются в единичном числе или исчезают вовсе. В случае дефектов ядерной ДНК их наследование может происходить от любого из родителей или от обоих родителей. Обычно при таких болезнях в митохондриях выявляются морфологические изменения.
Источник: medicalplanet.su
Организация дыхательной цепи транспорта электронов клеток эукариот
Стандартные окислительно-восстановительные потенциалы
Поскольку каждый переносчик электронов в электрон-транспортной цепи может попеременно выступать в роли, как восстановителя, так и окислителя, то он называется окислительно-восстановительным компонентом.
1) !!! Все окислительно-восстановительные компоненты реакции отличаются по своему сродству к электронам, т. е. способности принимать электроны.
2) Относительную способность веществ (участников электрон-транспортной цепи) принимать электроны (окисляться) выражают с помощью стандартного окислительно-восстановительного потенциала Е° (измеряется в вольтах).
3) Значение каждого окислительно-восстановительного потенциала измеряется по отношению к стандартному окислительно-восстановительному потециалу водорода при pH равному 7.
4) Условлено, что
а) — положительное значение (+) приписывается компоненту, обладающему большей способностью к восстановлению, чем водород;
б) — отрицательное значение приписывается компоненту, обладающему меньшей способностью к восстановлению, чем водород.
5) Любое соединение может отдавать электроны только соединению с более высоким окислительно-восстановительным потенциалом.
6) В дыхательной цепи каждое последующее звено имеет более высокий окислительно-восстановительный потенциал, чем предыдущее.
7) Процесс передачи электронов от одного окислительно-восстановительного компонента к другому, участвующих в превращении, характеризуют с помощью разности стандартных окислительно-восстановительных потенциалов ∆Е°.
8) Так же как и для значений разности стандартной свободной энергии ∆G°, в биохимии принято пользоваться значениями разности стандартных окислительно-восстановительных потенциалов (∆Ű´), полученными для стандартного состояния, соответствующего рН 7.
В клетках эукариот дыхательная цепь локализована во внутренней мембране митохондрий.
Митохондрии — это органеллы размером с бактерию (около 1 х 2 мкм). Обычно в клетке содержится около 2000 митохондрий, общий объем которых составляет до 25% от общего объема клетки (рис. 8.2):
Рис. 8.2. Схема строения митохондрии в разрезе
Митохондрия ограничена двумя мембранами — гладкой внешней искладчатой внутренней, имеющей очень большую поверхность. Складки внутренней мембраны глубоко входят в матрикс митохондрий, образуя поперечный перегородки — кристы. Пространство между внешней и внутренней мембраной носит название межмембранного пространства.
Внутренняя мембрана митохондрий характеризуется необычно высоким содержанием белков (75%). В их число входят транспортные белки-переносчики, ферменты, компоненты дыхательной цепи иАТФ-синтаза.
Источник: studopedia.su
Дисфункция митохондрий при нефропатиях у детей (Обзор литературы)
Ершова С. А.
Московский научно-исследовательский институт педиатрии и детской хирургии МЗ РФ, г. Москва Адрес для переписки: г. Москва, ул.Талдомская, д. 2. Московский НИИ педиатрии и детской хирургии МЗ РФ Телефон: 483-21-92
Ключевые слова: митохондриальные болезни, почки, диагностика, лечение
Введение
Достижения медицинской науки в области биохимии, клинической морфологии и медицинской генетики последних десяти лет позволили выделить новый гетерогенный пласт наследственных болезней у детей, обусловленных нарушением структуры и функций митохондрий и, как следствием, энергетической недостаточностью клеток. Митохондриальные нарушения — это обширная группа патологических состояний, связанных с дефектами митохондриального или ядерного генома [31]. Биохимические изменения, обусловленные дефектами митохондрий, проявляются нарушением транспорта митохондриальных субстратов, патологией утилизации субстратов, дефектами дыхательной цепи и недостаточным накоплением и передачей энергии [24]. Перечень митохондриальных болезней в последние годы неуклонно растет. Стали известны не менее 15 форм заболеваний, связанных с наследственными нарушениями транспорта и окисления жирных кислот, доказан их существенный вклад в происхождение гипогликемических состояний у детей, синдрома внезапной смерти [14]. Митохондриальные болезни трудны для диагностики в силу неспецифичности отдельных клинических проявлений, требуют разработки и внедрения новых программ диагностики, основанных на клинических, биохимических, молекулярно-генетических и морфологических критериях.
Впервые наблюдал митохондрии в виде гранул в мышечных клетках Kelliker в 1850 г. Позднее, в 1898 г., Mixaelis доказал, что митохондрии играют важную роль в окислительно-восстановительных процессах и клеточном дыхании.
30-х годов XIX века начато активное изучение структуры и функций митохондрий: была описана система цитохромов и окислительного фосфорилирования, G. Krebs разработал концепцию цикла трикарбоновых кислот. Новый прорыв в морфологическом и цитогенетическом аспектах изучения митохондрий был сделан в 60-е годы XX века, когда были открыты митохондриальный геном и митохондриальная ДНК. Возникла теория происхождения митохондрии: предполагалось, что в процессе филогенеза митохондрия как бактерия-симбионт встроилась в клетку. В 1963 г. Engel и Cunningham описали морфологический субстрат повреждения митохондрий — «рваные» красные волокна. В это же время R. Luft описал пациентку с нетиреоидным гиперметаболизмом, у которой было выявлено нарушение процессов окислительного фосфорилирования. Автор первым доказал прямую взаимосвязь дефекта митохондрий и появления патологических клинических проявлений. Последующие исследования XX-XXI веков были направлены на описание клинических симптомов и выявление новых нозологических форм митохондриальных болезней. Проводились дальнейшие исследования структур и функций митохондрий с помощью новейших технологий. При внедрении в клиническую практику методов молекулярной генетики была создана генетическая карта митохондриальных болезней. Это послужило основанием для разработки новых дифференцированных патогенетических подходов к терапии.
Структурно-функциональная характеристика митохондрий
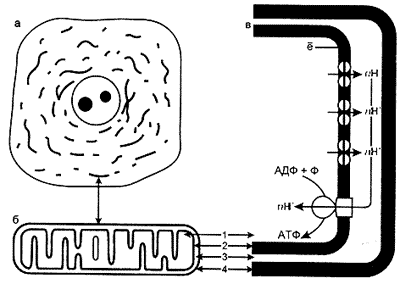
Митохондрия — внутриклеточная органелла, продуцирующая АТФ и содержащая уникальный геном, наследуемый в основном по материнской линии (неменделевское наследование) [79]. Митохондрии содержатся в цитоплазме всех аэробных эукариотических клеток [4]. По данным электронной микроскопии типичная митохондрия имеет форму короткого изогнутого цилиндра с закругленными концами. Ее длина около 1,5 мкм, диаметр 0,5 мкм. Митохондрии имеют двухслойную оболочку: наружная мембрана образует гладкую поверхность, на ней сконцентрированы ферменты, участвующие в транспорте и активации жирных кислот. От внутренней мембраны отходят многочисленные складки-кристы, где фиксируются комплексы ферментов, участвующих в переносе электронов и окислительном фосфорилировании (комплекс цитохромов В, С, А, А3) (рис.1).
Рис. 1. Строение и работа митохондрий:
а — митохондрии (указаны стрелкой), видимые в световом микроскопе;
б — ультраструктура митохондрий: 1 — митохондриальный матрикс, 2 — внутренняя митохондриальная мембрана, 3 — межмембранное пространство, 4 — внешняя митохондриальная мембрана;
в — общая схема функционирования митохондрий: при переносе электронов в цепи окисления в межмембранном пространстве накапливаются протоны и при достижении определенного потенциала возвращаются в матрикс; энергия этого потенциала тратится на синтез АТФ
Имеются сведения, что в клетках формируются и функционируют объединенные системы митохондрий, осуществляющих энергообеспечение тканей [1].
Основной функцией митохондрий является аэробное биологическое окисление (тканевое дыхание), в процессе которого идет высвобождение энергии и перенос протонов на свободные радикалы кислорода с образованием воды [11]. Другая важнейшая особенность организма человека и животных — способность накапливать выделяющуюся энергию в виде макроэргических соединений (АТФ, креатинфосфат и др.). Накопленная энергия в последующем трансформируется в механическую (в мышечных клетках), биоэлектрическую (в нервных клетках), в энергию активного транспорта (в клетках канальцевого эпителия почек).
Неотъемлемым свойством биологического окисления является сопряжение тканевого дыхания и окислительного фосфорилирования [11]. Установлено, что некоторые компоненты дыхательной цепи (коэнзим Q, цитохромоксидаза) наряду с переносом электронов по цепи осуществляют также перенос протонов из матрикса митохондрий в межмембранное пространство, в результате образуется протоновый градиент. В процессе обратного тока протонов внутрь митохондриального матрикса происходит утилизация освобождаемой в дыхательной цепи энергии путем фосфорилирования АДФ в АТФ и другие макроэргические фосфаты, создается запас энергии биологического окисления. Помимо транспорта электронов, окислительного фосфорилирования, митохондрии обеспечивают еще один процесс с вовлечением окислительно-восстановительных реакций — b-окисление жирных кислот. Свободные жирные кислоты трансформируются в ацетил-СоА и затем образуют эфиры с карнитином. Карнитин-ацетил-СоА переносится через митохондриальную мембрану, ацетил-СоА высвобождается и участвует в b-окислении.
Важной функцией митохондрий также является контроль за синтезом группы транспортных и рибосомальных РНК
Митохондриальная ДНК является небольшой двунитчатой молекулой, состоящей из тяжелой и легкой цепи, содержащей 16569 пар нуклеотидных оснований, 37 генов и имеющей собственный аппарат репликации [65]. Большинство митохондриальных белков кодируется ядерной ДНК, и лишь 2% — синтезируются в митохондриальном матриксе под контролем структурных генов. 13 генов отвечают за полипептиды дыхательной цепи: 7 относятся к комплексу I, 1 — к комплексу III, 3 — к комплексу IV, 2 — к комплексу V. Митохондриальная ДНК кодирует также 22 транспортные РНК и 2 рибосомальные РНК (12s, 16s). Остальные 70 полипептидов, входящих в состав I-V комплекса, кодируются ядерными генами, транспортируются в митохондрии и там функционируют [26].
Практически каждая клетка включает сотни и тысячи митохондрий, каждая из которых содержит от 2 до 10 молекул митохондриальной ДНК [38]. Причины, приводящие к мутациям митохондриальной ДНК, могут быть довольно разнообразны. Одними из самых распространенных — эндогенных — являются ошибки функционирования ДНК-полимераз и репараз (ферментов синтеза митохондриалъного генома) [8]. Другой механизм мутаций — это повреждение незащищенного гистонами и нитронами митохондриального генома продуктами перекисного окисления (супероксидные радикалы, перекись водорода, гидроксильные радикалы), так как митохондрии используют до 90% клеточного кислорода. Мутации митохондриальной ДНК представляют собой различного размера делеции (в том числе множественные), точечные поражения, деплеции, вставки. Репликация митохондриальной ДНК идет очень интенсивно (в 10 раз быстрее ядерной), происходит быстрое накопление мутаций. Многие факторы внешней среды (гипоксия, физические нагрузки, ионизирующее излучение) и лекарственные препараты также играют роль в патогенезе митохондриальных мутаций.
Митохондриальные расстройства могут возникать из-за нарушений либо в ядре, либо в митохондриальном геноме. Как результат может получиться любой вариант наследования: аутосомно-доминантный, аутосомно-рецессивный, Х-сцепленный, материнское наследование, спорадические случаи. Передача митохондрий и митохондриальной ДНК в следующее поколение в подавляющем большинстве случаев происходит через цитоплазму яйцеклетки [36].
Наличие большого числа копий митохондриальной ДНК в каждой клетке и их случайное распределение при клеточном делении определяют феномен гетероплазмии. Когда нормальный или мутантный геном всех митохондрий идентичен, клетка считается гомоплазмической. При наличии мутантной митохондриальной ДНК способность клеток осуществлять окислительное фосфорилирование определяется природой мутации, соотношением нормальных и мутантных геномов. Превышение нормального порога функционирования сопровождается нарушением энергетики и появлением клинических расстройств. Пороговый эффект зависит от различных факторов, в том числе от возраста и энергетической потребности ткани. Феномен гетероплазматической клетки объясняет наблюдения, что симптомы поражения того или иного органа могут усилиться или ослабнуть в процессе наблюдения, в то время как непораженные первоначально органы могут оказаться вовлеченными в патологический процесс.
Количество митохондрий контролируется посредством аутофагии. Старые, «изношенные» митохондрии уничтожаются лизосомами. Если по какой-либо причине количество митохондрий становится ниже необходимого и выработка ими АТФ уменьшается, то в клетке включаются другие механизмы получения энергии, такие, как процесс гликолиза в цитоплазме с выработкой небольшого количества АТФ и увеличением продукции молочной кислоты [15].
Табл. 1. Структурно-функциональная характеристика комплексов дыхательной цепи
|
Комплекс |
Функция |
Состав |
|
I (НАДН-коэнзим Q редуктаза) |
Транспорт электронов от НАДН2-продуцирующих субстратов на убихинон |
25-28 полипептндов, 7 из которых кодируются митохондриальной ДНК |
|
II (сукцинат-коэнзим Q редуктаза) |
Перенос редуцирующих эквивалентов от ФАДН2-продуцирующих субстратов на убихинон (коэнзим Q10) |
5 ядернокодируемых пептидов |
|
III (коэнзим Q цитохром С редуктаза) |
Транспорт электронов от восстановленного убихинона |
11 субъединиц, 1 кодируется митохондриальной ДНК |
|
IV (цитохром С оксидаза) |
Окончательный этап передачи электронов от восстановленного цитохрома С на молекулярный кислород. Поддержка активности протонного насоса |
Цитохромы А, А3 и 13 протеиновых субъединиц, 3 из которых кодируются митохондриально |
|
V (АТФ-синтетаза) |
Обеспечение обратного тока протонов внутрь митохондриального матрикса, использование освободившейся энергии на синтез АТФ |
|
Диагностика митохондриальных заболеваний
Современная клиническая диагностика митохондриальной патологии требует проведения поиска с использованием самой сложной технологии — хромато-масс-спектрометрии, электронно-оптического, гистохимического исследований биопсийного материала и т.д.
Обследование пациентов с подозрением на митохондриальную цитопатию включает: исследование уровней лактата, пирувата, кетоновых тел и их соотношение натощак и после различных нагрузок, определение аминокислотного состава крови и мочи, определение содержания жирных кислот, спектра липидов и фосфолипидов, общего и свободного карнитина и продуктов перекисного окисления липидов в крови [22].
Доступными и информативными для динамической оценки интенсивности аэробных окислительных процессов в организме оказались цитохимические тесты на активность сукцинатдегидрогеназы лимфоцитов периферической крови (количественный цитохимический метод, модифицированный Нарциссовым, 1969), а также других митохондриальных ферментов: лактатдегидрогеназы, глицерофосфатдегидрогеназы. Морфометрическими методами удалось показать, что функциональная активность митохондрий лимфоцитов у детей с митохондриальными болезнями достоверно снижена на 11-23% по сравнению со здоровыми детьми. Р.П. Нарциссовым и B.C. Сухоруковым получены данные о достоверной корреляции функциональной активности митохондрий лимфоцитов, отражающие полисистемную митохондриальную недостаточность с тестом на «рваные» красные волокна в биоптатах скелетных мышц [19]. Кроме того, цитохимический метод применим для оценки эффективности применяемой терапии, так как изменение функциональной активности митохондрий после приема препаратов, например содержащих янтарную кислоту, отмечается уже через 40 минут после введения лекарства.
Характерная клиническая картина и/или метаболические изменения, предполагающие наличие митохондриальной цитопатии, требуют непосредственного исследования митохондриальной дыхательной цепи при помощи полярографических и полярометрических исследований митохондрий, полученных при биопсии [24,65].
Характерным гистологическим признаком митохондриальных расстройств при световой микроскопии с применением различных методов окраски служат: наличие «рваных» (шероховатых) красных волокон (RRF) в биоптатах мышц [32], накопление гликогена и липидов, атрофия, вакуолизация и перераспределение волокон 1-го и 2-го типов, дефицит митохондриальных ферментов цикла Кребса и дыхательной цепи (цитохром С оксидазы, сукцинатдегидрогеназы, НАД*Н-оксиредуктазы и др.), некрозы единичных или мелких групп мышечных волокон, эпителиальных клеток [8,13].
При электронной микроскопии обнаруживаются изменения, характерные для глубоких повреждений митохондрий:
-
значительные скопления органелл под сарколеммой;
-
полиморфизм с наличием крупных, удлиненных, почкующихся форм и/или преобладание мелких овальных органелл;
-
дезорганизация архитектоники и структуры крист, их набухание, фрагментация, утрата;
-
наличие депозитов кальция, кристаллических включений;
-
лизис органелл с участием лизосом, образованием мембранных телец;
-
снижение активности митохондриальных ферментов.
Клиническая картина митохондриальных заболеваний
Дисфункция митохондрий у детей развивается двояко — в форме симптомокомплекса установленной митохондриалыюй природы или в виде гетерогенной патологии, при которой не исключаются варианты вторичной митохондриальной недостаточности. Разработка классификации митохондриальных болезней начата сравнительно недавно и активно продолжается, предлагаются различные принципы классификации: клинический, генетический, биохимический. В настоящий момент используется генетическая классификация митохондриальных болезней, предложенная D.de Vivo в 1993 г.
-
Врожденные (наследственные)
-
Дефекты ядерной ДНК:
-
дефекты транспортных субстратов;
-
дефекты субстратов утилизации;
-
дефекты цикла Кребса;
-
нарушение окислительного фосфорилирования;
-
нарушения в дыхательной цепи;
-
дефекты импортирования белков.
-
-
Дефекты митохондриальной ДНК:
-
спорадические р-мутации;
-
точечные мутации с поражением структурных генов;
-
точечные мутации синтезирующих генов.
-
-
Межгеномные сигнальные дефекты:
-
миделеции митохондриальной ДНК, множественные аутосомно-доминантные;
-
делеции митохондриальной ДНК, множественные аутосомно-рецессивные.
-
-
-
Приобретенные: недостаточность митохондрий, обусловленная действием:
-
токсинов, экопатогенов;
-
лекарственных препаратов;
-
старением.
-
К настоящему времени верифицированы самостоятельные нозологические формы, возникающие при мутациях митохондриальных генов, к ним относят: синдромы, такие, как MELAS (энцефалопатия, лактат-ацидоз, инсультоподобные эпизоды), MERRF (миоклонус-эпилепсия, красные «рваные» волокна), Кернса-Сейра (пигментный ретинит, атаксия, офтальмоплегия, мышечная слабость, нарушение сердечной проводимости), Пирсона (вялость, гипопластическая анемия, нарушение функций поджелудочной железы, диарея), оптическая нейропатия Лебера, синдром DIDMOAD (сахарный и несахарный диабет, атрофия зрительных нервов, нейросенсорная тугоухость), NARF (атаксия, нейропатия, пигментный ретинит).
Выделена также группа митохондриальных болезней, связанных с ядерными мутациями, среди них различные формы младенческих миопатий, болезни Альперса — прогрессирующая энцефалопатия (дегенерация серого вещества мозга в сочетании с циррозом печени), Лея (подострая невротизирующая энцефаломиелопатия, мышечная гипотония, атаксия и нистагм, пирамидные симптомы, офтальмоплегия и атрофия зрительных нервов, часто отмечается присоединение кардиомиопатий), Менкенса (резкая задержка психомоторного развития, отставание в росте, нарушение роста и дистрофические изменения волос), синдром Барта (задержка физического и психомоторного развития, миопатический синдром, гипертрофическая кардиомиопатия, нейтропения, гипогликемические состояния), синдромы недостаточности карнитина, некоторых ферментов цикла Кребса и дыхательной цепи. В этом случае тип наследования заболевания соответствует простой менделевской передаче по аутосомно-доминантному или аутосомно-рецессивнохму типу.
Митохондриальные заболевания могут встречаться в любом возрасте, однако у одной трети пациентов с недостаточностью ферментов дыхательной цепи начальные симптомы проявляются в первый месяц жизни. Двумя основными клиническими признаками митохондриальных расстройств являются увеличение с течением времени числа вовлеченных в патологический процесс органов и тканей, а также практически неизбежное поражение центральной нервной системы [29].
Имеется значительная вариабельность симптомов, наличие стертых и скрытых форм. У детей с дисфункцией митохондрий часто выявляются: отставание физического развития, сниженная масса тела (инфантильный соматотип), гипотония скелетных мышц, астения, нарушение терморегуляции, обморочные состояния [53]. Так называемые «вялые дети» нередко имеют митохондриальную недостаточность.
Изначальный взгляд на митохондриальные болезни как на нервно-мышечную патологию сформировался не случайно: поражение ЦНС и мышечной ткани доминирует в клинике. Миопатический синдром включает в себя слабость и атрофию проксимальной мускулатуры, мышечные боли, непереносимость физической нагрузки, прогрессирующую наружную офтальмоплегию, птоз, отсутствие рефлексов. Основными неврологическими проявлениями являются судороги, инсульты (инсультоподобные эпизоды), нейросенсорная тугоухость, атрофия зрительного нерва, атаксия, миоклонусы, крампи, полинейропатия, олигофрения, деменция, нарушение психомоторного развития, мигрень [9,20,31]. Митохондриальные изменения являются клинико-патогенетической основой для развития кардиомиопатий (идиопатическая дилатационная и симметричная гипертрофическая кардиомиопатий, кардиомиопатии при синдромах Кернса-Сейра, MELAS, Барта) [17], нарушения проводимости (сердечные блокады). Клетки миокарда в условиях митохондриальной недостаточности изменяются принципиально также, как и волокна скелетных мышц, что подтверждается наличием феномена «рваных» красных волокон в сердечной мышце [66].
Среди эндокринопатий, выявленных при митохондриалыюй патологии, первое место занимает сахарный диабет [43,61]. Также описаны гипопаратиреоз, изолированный дефицит гормона роста, гипогонадизм, экзокринная недостаточность поджелудочной железы при синдроме Пирсона [62].
Патология желудочно-кишечного тракта проявляется в виде появления повторной рвоты (особенно после физической нагрузки), диареи или псевдонепроходимости кишечника [69]. Серьезное поражение печени наблюдается при ряде синдромов с первичным поражением митохондриальной ДНК, при митохондриальной патологии с преимущественным нарушением b-окисления жирных кислот (например стеатоз печени при синдроме Рея). В условиях первичного биллиарного цирроза печени выявляется деструкция желчных протоков с наличием гетерогенных клонов Т-лимфоцитов в инфильтратах портальных трактов. Поражение печени характеризуется прогрессирующим увеличением печени с нарушением функций и развитием признаков печеночной недостаточности [27].
Описано поражение костного мозга с развитием панцитопении у новорожденных, трансфузиозависимой макроцитарной анемии, тромбоцитопении, нейтропении в старшем возрасте при синдроме Пирсона [48].
Патология почек
Исследование митохондриальных нарушений, их вклад в развитие патологического процесса в мировой нефрологии и урологии проводятся в течение 5 лет. Е.Л. Вишневский [3] показал роль митохондриальных нарушений в развитии нейрогенной дисфункции мочевого пузыря, а также в прогнозировании развития энуреза [21]. Проводились работы по изучению энергетической недостаточности при дисфункциях почечной лоханки и мочеточников у детей с гидронефротической трансформацией почек [16]. О.В. Комарова, Т.В. Сергеева определили снижение функциональной активности митохондриальных ферментов в лимфоцитах у детей с хроническим гломерулонефритом при воздействии пульс-терапии стероидами [10].
Ультраструктура нефрона такова, что наиболее богатые митохондриями клетки находятся в проксимальных и дистальных извитых канальцах коркового слоя почки, а также в восходящей части петли Генле, лежащей в наружной части мозгового вещества. В связи с этим диагностическим ориентиром почечной митохондриальной дисфункции, прежде всего, служит нарушение деятельности именно этих структур нефрона, что важно для ранней диагностики патологии. Поражение митохондрий различных отделов почек может быть как первичным (в рамках митохондриальных цитопатий), так и вторичным: в результате различных почечных заболеваний и токсических воздействий тяжелых металлов и некоторых лекарственных средств.
Выделено четыре основных клинико-морфологических варианта нефропатий с митохондриальной дисфункцией:
-
тубулопатии с поражением эпителия проксимальных и (или) дистальных канальцев;
-
тубулоинтерстициальные нефриты [68];
-
смешанные нефропатии, проявляющиеся фокально-сегментарным гломерулосклерозом [63];
-
дизметаболические нефропатии (чаще с оксалано-кальциевой кристаллурией).
В отдельную группу вынесена патология мочевого пузыря, связанная с митохондриальными дисфункциями.
Тубулопатии с поражением эпителия проксимальных и (или) дистальных канальцев
Самым ранним признаком проявления заболевания почек, вызванным первичной митохондриальной недостаточностью, является триада Фанкони и болезнь де Тони-Дебре-Фанкони [64]. Этиологическим фактором в данном случае могут быть дефекты пируватдегидрогеназного комплекса [44] и нарушения в дыхательной цепи на уровне III комплекса (коэнзим Q цитохром С редуктаза) и IV комплекса — ключевого элемента дыхательной цепи — цитохром С оксидазы [56]. Клинически синдром де Тони-Дебре-Фанкони, вызванный мутациями в митохондриях, проявляет себя, как и в классическом варианте, в виде проксимальной канальцевой недостаточности: глюкозурии, генерализованной гипераминоацидурии, фосфатурии, тубулярной протеинурии, гипокалиемии, гипоурикемии [57,71]. Эти симптомы обычно возникают в раннем неонатальном периоде и приводят к развитию терминальной почечной недостаточности [25]; либо у детей до 3 лет, причем развитие проявлений сложной канальцевой недостаточности может на несколько лет предшествовать типичным проявлениям митохондриальной энцефаломиопатии [52], в том числе и синдромам Кернса-Сейра и Пирсона [33,35]. Потеря воды и натрия приводит к появлению полиурии (до 2-3 литров в сутки), полидипсии, склонности к гипертермии и кризам обезвоживания. Из-за нарушения реабсорбции бикарбонатов развиваются признаки ацидоза и постепенно прогрессирующие изменения со стороны костной системы (рахитоподобные изменения скелета, задержка роста, спонтанные переломы). В почечных биоптатах на этом этапе патологии определяются белковая дистрофия канальцевых нефроцитов, атрофия и дедифференцировка эпителия с нарушением контактов между эпителиоцитами и умеренное расширение просвета проксимальных или дистальных канальцев нефрона и петли Генле, закупорка их цилиндрами. В некоторых клетках выявлены вакуолизация цитоплазмы и наличие гигантских митохондрий, что связано с угнетением митохондриального дыхания, нарушением окислительного фосфорилирования [71]. В литературе есть данные о возникновении нефропатий с развитием канальцевой дисфункции — синдрома де Тони-Дебре-Фанкони в результате повреждения митохондрий при острой свинцовой интоксикации [49].
Другими тубулярными нарушениями, описанными при первичных митохондриальных цитопатиях, являются дистальный почечный канальцевый ацидоз, тубулопатия, подобная синдрому Барттера [37], и проксимальный почечный канальцевый ацидоз (причина — парциальный дефицит цитохром С оксидазы [49]) при синдроме Кернса-Сейра, а также при синдроме Пирсона [56], для которого характерны гиперкальциурия и гипероксалурия.
Приведено описание больных с недостаточностью цитохром С оксидазы, фенотип которых был идентифицирован как подострая некротизирующая энцефаломиелопатия Лея с клиническими проявлениями почечного тубулярного ацидоза [12].
В литературе имеются сведения о развитии кист при митохондриальных тубулопатиях. Дисфункция митохондрий эпителия почечных канальцев и связанная с ней слабость клеточной энергетики способствуют, вероятно, ухудшению белкового синтеза и ослаблению межклеточных контактов со склонностью к образованию кист в том органе, патология которого имеет яркие проявления, в частности в почке [7]. Множественные кисты коркового вещества описаны при синдроме Пирсона [34]. Комбинация поликистоза почек и жировой дистрофии печени может явиться следствием глютаровой ацидурии II типа — аутосомно-рецессивного дефекта энергетического метаболизма митохондрий, приводящего к летальному исходу в неонатальном периоде [46].
Тубулоинтерстициальные нефриты
Тубулоинтерстициальные нефриты у больных с митохондриальными цитопатиями чаще других нефропатий связаны с возможным неблагоприятным экзогенным воздействием [7]. Заболевание проявляется хронической почечной недостаточностью без признаков дисфункции проксимальных канальцев [73], у 6 описанных больных отмечалась полиурия со снижением контрационной функции почек и экстраренальными изменениями [63]. Световая микроскопия почечных биоптатов при хроническом тубулоинтерстициальном нефрите выявляет диффузный интерстициальный фиброз с тубулярной атрофией и склерозом гломерул в зоне почечного фиброза. Методом электронной микроскопии обнаруживаются признаки явной деэнергизации митохондрий эпителия извитых канальцев нефрона: дистопия митохондрий (они располагались либо хаотически, без связи с инвагинациями цитомембраны, либо группировались около ядра), дезорганизация интрамитохондриальных элементов (уплотненное расположение крист, уплотнение матрикса, разрыв крист, формирование интрамитохондриальных пластинчатых структур типа миелиноподобных и др. — рис.2). Особо четко наблюдаемые патологические изменения регистрировались в нефробиоптатах у детей с признаками дизэмбриогенеза [13].
Источник: StudFiles.net